
TỔNG QUAN VỀ PHOSPHOLIPID
27/02/2025
Định lượng protein bằng TMT – Tổng quan và ứng dụng
03/03/2025
Phương pháp xử lý dữ liệu Proteomics
Phân tích dữ liệu proteomics là quá trình xử lý và diễn giải các tập dữ liệu lớn được tạo ra từ nghiên cứu về protein, bao gồm cấu trúc, chức năng và tương tác của chúng trong một hệ thống sinh học. Proteomics và post-translational modification (PTM) omics đã tìm thấy các ứng dụng rộng rãi trong khoa học sự sống, y học cơ bản và nhiều lĩnh vực nghiên cứu khác. Tuy nhiên, giống như các dữ liệu đa omics khác, dữ liệu proteomics và omics PTM đòi hỏi phải kiểm soát chất lượng và xử lý nghiêm ngặt trước khi phân tích chính thức. Nhìn chung, quy trình xử lý trước cho dữ liệu proteomics và PTM omics bao gồm một số bước chính:
- Thu thập dữ liệu thô: Thu thập dữ liệu ban đầu thông qua kỹ thuật phổ khối.
- Tìm kiếm cơ sở dữ liệu: Sử dụng cơ sở dữ liệu để đối chiếu quang phổ thu được với trình tự protein đã biết.
- Định lượng: Đo mức độ phong phú của protein.
- Biến đổi logarit: Ứng dụng biến đổi logarit để ổn định phương sai.
- Chuẩn hóa: Điều chỉnh dữ liệu theo một thang đo chung, cho phép so sánh có ý nghĩa.
- Xử lý giá trị bị thiếu: Xử lý và nhập các giá trị bị thiếu trong tập dữ liệu.
- Loại bỏ hiệu ứng theo lô: Giảm thiểu các biến thể có hệ thống do các lô thử nghiệm khác nhau gây ra.
- Phân tích biểu hiện khác biệt: Xác định và khám phá các protein biểu hiện sự khác biệt đáng kể.
Các phòng thí nghiệm, công nghệ proteomics, công cụ phần mềm và thậm chí cả mô khác nhau có thể biểu hiện những thay đổi đáng kể trong các phương pháp tiền xử lý dữ liệu proteomics. Dựa trên bối cảnh thử nghiệm cụ thể và mục tiêu nghiên cứu, các phương pháp tiền xử lý cần được nhấn mạnh tầm quan trọng về quy trình làm việc đã chọn. Một số quy trình tiền xử lý thường được sử dụng trong proteomics:
Quy trình xử lý dữ liệu DIA Proteomics
Trong phương pháp DIA, các ion tiền chất được phân lập thành các cửa sổ phân lập được xác định trước và phân mảnh; tất cả các ion phân mảnh trong mỗi cửa sổ sau đó được phân tích bằng máy quang phổ khối có độ phân giải cao. Trong lĩnh vực DIA (Data-Independent Acquisition) proteomics, việc xử lý trước dữ liệu đóng vai trò then chốt trong việc đảm bảo độ tin cậy và độ chính xác của kết quả nghiên cứu. Hiện nay, có hai quy trình xử lý trước dữ liệu nổi bật đang thịnh hành: chiến lược tìm kiếm cơ sở dữ liệu Spectronaut và chiến lược tìm kiếm cơ sở dữ liệu DIA-NN.
- Chiến lược tìm kiếm cơ sở dữ liệu Spectronaut
Spectronaut có thể định lượng hồ sơ từ 100 đến vài nghìn protein trong một thí nghiệm. Có thể phân tích các thí nghiệm lớn với nhiều điều kiện và bản sao bao gồm tới 10.000 lần chạy LC-MS. Một nghiên cứu đáng chú ý minh họa cho chiến lược Spectronaut là công trình do nhóm nghiên cứu của Matthias Mann thực hiện, được công bố trên “Nature Medicine” năm 2022 (PMID: 35654907). Nghiên cứu này tập trung vào dữ liệu proteomics của DIA có nguồn gốc từ mô và huyết tương, được xử lý bằng Spectronaut v13 và v15.4 (Hình 1). Quy trình làm việc bắt đầu bằng việc thu thập thông tin định lượng tương đối, sau đó là chuyển đổi log2. Sau đó, dữ liệu được xử lý đối với các giá trị bị thiếu, với các nhà nghiên cứu lựa chọn phương pháp tính toán bằng cách sử dụng các giá trị ngẫu nhiên được rút ra từ phân phối chuẩn. Dữ liệu được tính toán sau đó được tiến hành phân tích thống kê và tin sinh học hạ lưu.
Điều đáng chú ý là nghiên cứu này không nêu rõ phương pháp chuẩn hóa dữ liệu và cách tiếp cận để giải quyết các hiệu ứng theo lô. Trong các nghiên cứu khác, thông lệ chung là thực hiện chuẩn hóa trung vị trong các mẫu sau khi chuyển đổi log2 và trước khi xử lý các giá trị bị thiếu. Bước này đảm bảo rằng dữ liệu từ các mẫu khác nhau được chuẩn hóa, tạo điều kiện thuận lợi cho việc so sánh protein khác biệt giữa các mẫu. Việc giảm thiểu các hiệu ứng theo lô thường đạt được bằng cách sử dụng phương pháp Combat đã được thiết lập tốt.
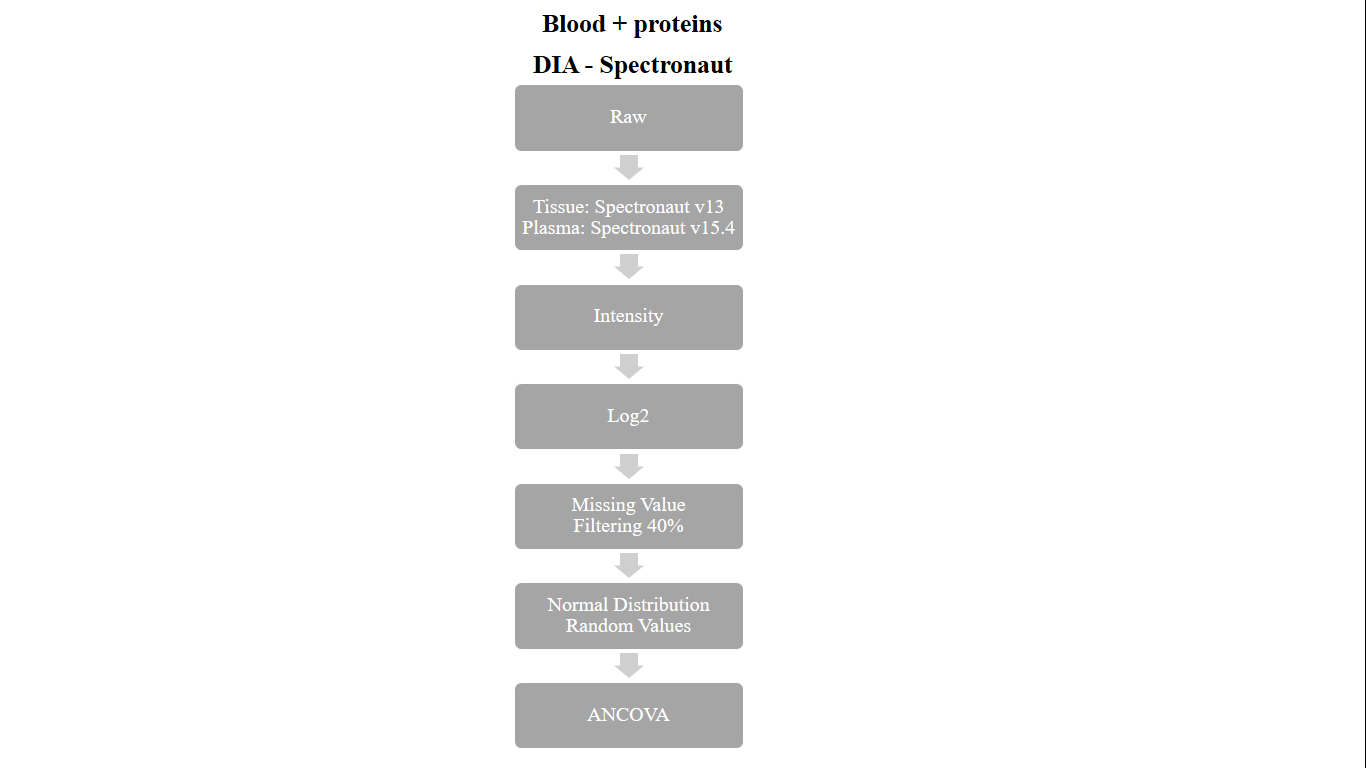
Hình 1. Quy trình xử lý trước chiến lược tìm kiếm cơ sở dữ liệu Spectronaut (PMID: 35654907) (Tâm Trần, Research Officer, Hoan Vu Biomolecules., JSC).
- Chiến lược tìm kiếm cơ sở dữ liệu DIA-NN
Chiến lược DIA-NN phản ánh cách tiếp cận của Spectronaut trong các bước xử lý dữ liệu trước. DIA-NN bao gồm một thuật toán để phát hiện và loại bỏ nhiễu từ phổ tandem-MS. Đối với mỗi đỉnh rửa giải, DIA-NN chọn đoạn ít bị ảnh hưởng nhất bởi nhiễu (là đoạn có hồ sơ rửa giải tương quan tốt nhất với hồ sơ rửa giải của các đoạn khác). DIA-NN có thể đọc các tệp mzML cũng như nhập trực tiếp dữ liệu thô từ các lần thu thập Sciex và Thermo. Để xử lý chúng, DIA-NN yêu cầu phải cung cấp thư viện phổ hoặc cơ sở dữ liệu trình tự làm đầu vào và DIA-NN tạo ra một thư viện phổ trong silico. Đối với điều này, DIA-NN có thể tùy chọn sử dụng một bộ dự đoán phân mảnh và một bộ dự đoán thời gian lưu tuyến tính. Các bộ dự đoán được đào tạo bằng bất kỳ thư viện phổ nào do người dùng cung cấp. Sau khi tìm kiếm cơ sở dữ liệu DIA-NN, dữ liệu trải qua một chuỗi các quy trình bao gồm chuyển đổi log2, chuẩn hóa dữ liệu và xử lý giá trị bị thiếu, kết thúc bằng việc xác định các protein được biểu hiện khác biệt.
Hình 2. Quy trình xử lý dữ liệu trước cho chiến lược tìm kiếm cơ sở dữ liệu DIA-NN (Tâm Trần, Research Officer, Hoan Vu Biomolecules., JSC).
Tóm lại, cả chiến lược Spectronaut và DIA-NN đều kết hợp các bước tiền xử lý tương đương trong các nghiên cứu proteomics của DIA. Tuy nhiên, các nhà nghiên cứu phải nhận thức được các cân nhắc bổ sung như chuẩn hóa dữ liệu và hiệu chỉnh hiệu ứng lô để đảm bảo tính mạnh mẽ và chính xác của các phân tích hạ nguồn. Việc khám phá sâu hơn và các nghiên cứu so sánh các phương pháp này sẽ góp phần tinh chỉnh và tối ưu hóa quy trình tiền xử lý dữ liệu trong proteomics của DIA.
Quy trình xử lý dữ liệu không nhãn
Quy trình xử lý trước cho dữ liệu proteomics không có nhãn khác với dữ liệu DIA (Thu thập dữ liệu độc lập) và chủ yếu liên quan đến hai phương pháp tìm kiếm cơ sở dữ liệu: MaxQuant và Proteome Discoverer.
» Kết quả tìm kiếm cơ sở dữ liệu MaxQuant
MaxQuant là một phần mềm phân tích dữ liệu proteomics phổ biến, được sử dụng để xử lý dữ liệu từ các thí nghiệm khối phổ (mass spectrometry) với nhiều bước phân tích:
- Phát hiện đỉnh và chấm điểm peptide: MaxQuant hiệu chỉnh những sai lệch có hệ thống về khối lượng peptide được đo và thời gian lưu tương ứng.
- Hiệu chuẩn khối lượng: Phát hiện khối lượng và cường độ của các đỉnh peptide trong quang phổ MS và lắp ráp chúng thành các đỉnh 3D trên mặt phẳng thời gian lưu m/z, sau đó lọc để xác định các mẫu đồng vị.
- Tìm kiếm cơ sở dữ liệu để nhận dạng protein: Khối lượng peptide và đoạn peptide (trong trường hợp phổ MS/MS) được tìm kiếm trong cơ sở dữ liệu trình tự cụ thể của sinh vật, sau đó được chấm điểm theo phương pháp dựa trên xác suất gọi là điểm peptide.
- Định lượng protein: Độ chính xác khối lượng cao đạt được bằng cách tính trung bình có trọng số và thông qua hiệu chuẩn khối lượng.
Khi thực hiện định lượng protein, MaxQuant cung cấp ba giá trị định lượng chính:
- Intensity: Giá trị cường độ tín hiệu (intensity) của peptide hoặc protein, phản ánh lượng tương đối của chúng trong mẫu. Cường độ này được tính toán dựa trên diện tích peak (peak area) trong phổ khối.
- LFQ (Label-Free Quantification): Đây là phương pháp định lượng không sử dụng nhãn (label-free), dựa trên việc so sánh cường độ tín hiệu giữa các mẫu khác nhau. LFQ cho phép định lượng tương đối protein giữa các mẫu mà không cần sử dụng các chất đánh dấu đồng vị (isotopic labels).
- iBAQ (Intensity-Based Absolute Quantification): iBAQ là phương pháp định lượng tuyệt đối dựa trên cường độ tín hiệu. Giá trị iBAQ được tính bằng cách chia tổng cường độ tín hiệu của tất cả các peptide của một protein cho số lượng lý thuyết của các peptide có thể quan sát được (theo trình tự protein). iBAQ giúp ước lượng lượng tuyệt đối của protein trong mẫu.
Đối với các phân tích định lượng tiếp theo, cho dù sử dụng Intensity, iBAQ hay LFQ, thì việc thực hiện chuyển đổi log2, chuẩn hóa trung vị theo mẫu hoặc chuẩn hóa phân vị là điều thường thấy. Sau đó, các giá trị bị thiếu được lọc và quy kết, dẫn đến bước cuối cùng là phân tích định lượng khác biệt.
Tìm kiếm cơ sở dữ liệu Proteome Discoverer
Trong Proteome Discoverer (PD), giá trị định lượng mặc định là iBAQ. Phương pháp chuẩn hóa thường được sử dụng là FOT (Fraction of Total), bao gồm chuẩn hóa dựa trên tổng giá trị iBAQ cho tất cả các protein trong một mẫu. Sau đó, các giá trị bị thiếu được tính toán dựa trên phân phối dữ liệu bằng các tham số như 10e -5 hoặc 10e -8 , giống như một dạng tính toán giá trị tối thiểu. Dữ liệu đã xử lý sau đó trải qua các quy trình phân tích hạ lưu.
» Quy trình xử lý dữ liệu TMT
Công nghệ proteomics TMT (Tandem Mass Tag) được sử dụng rộng rãi, đặc biệt là trong các nghiên cứu mẫu lớn và nhóm lâm sàng. TMT sử dụng các chất đánh dấu (tags) hóa học để gắn vào các nhóm amin của peptide hoặc protein trong các mẫu khác nhau. Các tags này có khối lượng phân tử khác nhau nhưng có tính chất hóa học tương tự, cho phép phân biệt các mẫu trong cùng một lần chạy máy khối phổ (mass spectrometry). Các phương pháp tìm kiếm cơ sở dữ liệu proteomics TMT rất đa dạng, bao gồm MaxQuant, Proteome Discoverer (PD), MSFragger và MS-GF+.
Lấy ví dụ về chiến lược tìm kiếm cơ sở dữ liệu MSFragger, quy trình xử lý trước cho dữ liệu nghiên cứu về protein TMT được minh họa như sau:
- MSFragger ban đầu thực hiện tìm kiếm cơ sở dữ liệu trên dữ liệu proteomics thô, tạo ra các tệp kết quả tìm kiếm có định dạng pepXML.
- Bộ công cụ Philosopher được sử dụng để định lượng và lọc peptide, protein và sửa đổi sau dịch mã (PTM). Cụ thể, đầu ra từ MSFragger có thể trải qua quá trình nhận dạng và xác thực peptide bằng PeptideProphet. Đối với các tập dữ liệu giàu sửa đổi như phosphoryl hóa, PTMProphet được sử dụng để nhận dạng tại các vị trí sửa đổi dựa trên kết quả PeptideProphet. Nhận dạng protein được xử lý bằng ProteinProphet.
- Cuối cùng, Philosopher được sử dụng để lọc tỷ lệ phát hiện sai ((False Discovery Rate-FDR) và định lượng, tạo ra cường độ ion báo cáo TMT ở mức peptide, sửa đổi hoặc protein. Tuy nhiên, giá trị định lượng tương đối cho tất cả các protein trong mỗi mẫu cần phải hiệu chỉnh dựa trên mẫu kênh tham chiếu. Điều này liên quan đến việc tính toán tỷ lệ (tỷ lệ TMT) của cường độ TMT cho một protein trong một mẫu nhất định so với cường độ của cùng một protein trong mẫu kênh tham chiếu.
- Sau khi thu thập các giá trị tỷ lệ TMT cho mỗi protein, một phép biến đổi log2 được áp dụng và một quy trình chuẩn hóa trung vị trong mẫu được tiến hành. Chuẩn hóa này không phải là phép chia đơn giản các giá trị tỷ lệ TMT cho trung vị; thay vào đó, nó bao gồm nhiều bước biến đổi dữ liệu. Ban đầu, tỷ lệ TMT trung vị cho mỗi mẫu được tính toán và trung vị toàn cục M0 được xác định. Sau đó, theo các chiến lược phân tích thông thường, mỗi mẫu trải qua quá trình chuẩn hóa trung vị dựa trên trung vị tương ứng của nó và độ lệch trung vị tuyệt đối (MAD) cho mỗi mẫu được tính toán. Độ lệch trung vị tuyệt đối toàn cục MAD0 cho tất cả các mẫu sau đó được xác định. Cuối cùng, các giá trị định lượng tương đối của protein được chuẩn hóa dựa trên M0 và MAD0.
- Giá trị biểu hiện tương đối cuối cùng (A) cho một protein được biểu diễn dưới dạng giá trị định lượng protein chuẩn hóa (sau khi chuyển đổi log2), được cộng vào giá trị tương ứng của protein kênh tham chiếu (sau khi chuyển đổi log2).
- Sau đó, tiếp theo là các quy trình như xử lý giá trị bị thiếu, loại bỏ hiệu ứng lô và phân tích biểu hiện khác biệt hạ lưu.
Tài liệu tham khảo
- Demichev V, Messner CB, Vernardis SI, Lilley KS, Ralser M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat Methods. 2020;17(1):41-44. doi:10.1038/s41592-019-0638-x
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367-1372. doi:10.1038/nbt.1511


