
Matrix effects and application of matrixeffect factor
03/03/2025
Applications of Tandem Mass Spectrometry (MS/MS) in Protein Analysis for Biomedical Research
05/03/2025
Kỹ thuật định lượng không nhãn
Kỹ thuật định lượng không nhãn là gì?
Ngày nay, các nghiên cứu về proteomics không còn chỉ tập trung vào việc xác định càng nhiều protein càng tốt trong một mẫu nhất định, mà hướng đến mục tiêu định lượng chính xác các protein đó. Việc nghiên cứu các hồ sơ biểu hiện protein biến đổi có thể mang lại thông tin hữu ích về các con đường bệnh lý hoặc các dấu ấn sinh học và các mục tiêu thuốc liên quan đến một căn bệnh cụ thể. Theo thời gian, nhiều phương pháp tiếp cận proteomics định lượng đã được thiết lập, cho phép các nhà nghiên cứu trong lĩnh vực proteomics tham khảo một bộ công cụ toàn diện gồm nhiều phương pháp khác nhau. Kỹ thuật định lượng không nhãn protein phân tích các mảnh peptide thu từ các protein được cắt mạch bằng enzyme dựa vào phương pháp LC chất lượng cao và so sánh cường độ tín hiệu của các mảnh peptide tương ứng trong các mẫu khác nhau để định lượng tương đối protein. Phương pháp này không phụ thuộc vào nhãn đồng vị và không yêu cầu nhãn đồng vị ổn định đắt tiền làm tham chiếu nội bộ. Kỹ thuật này hiện được sử dụng rộng rãi trong các lĩnh vực sàng lọc dấu hiệu bệnh, nghiên cứu cơ chế phát triển bệnh và nghiên cứu mục tiêu tác dụng của thuốc1.
Proteomics không nhãn đã nổi lên như một phương pháp thông lượng cao cho các nghiên cứu proteomics lâm sàng định lượng. Các phương pháp định lượng proteomics không nhãn có thể được chia thành hai chiến lược định lượng khác nhau.
- Phương pháp đầu tiên được gọi là đếm phổ và ngụ ý việc đếm và so sánh số phổ ion phân mảnh (MS/MS) thu được đối với các peptide của một protein nhất định2. Các phương pháp tiếp cận đã sửa đổi của đếm phổ đã được báo cáo, có tính đến các khía cạnh ảnh hưởng đến số lần đếm phổ, như các tính chất lý hóa của peptide cũng như độ dài của các protein tương ứng. Những cách tiếp cận này được gọi là biểu hiện protein tuyệt đối (APEX)3 và hệ số phong phú phổ chuẩn hóa (NSAF)4.
- Cách tiếp cận thứ hai của proteomics định lượng không nhãn ngụ ý việc đo diện tích đỉnh sắc ký (còn được gọi là cường độ tín hiệu khối phổ) của các ion tiền chất peptide. Tùy thuộc vào phương pháp sắc ký (ví dụ: sắc ký lỏng pha đảo ngược), các peptide được tách ra theo các tính chất vật lý cụ thể của chúng (ví dụ: kỵ nước, điện tích), sau đó là nguồn anion ion hóa và cuối cùng được phát hiện trong máy quang phổ khối. Trong phổ khối lượng thu được, mỗi peptide có điện tích và khối lượng cụ thể tạo ra một đỉnh khối lượng đơn đồng vị. Cường độ của đỉnh này theo thời gian lưu có thể được hình dung trong sắc ký đồ ion chiết xuất (XIC) và diện tích dưới đường cong (AUC) có thể được xác định. Diện tích của các đỉnh sắc ký đã được chứng minh là có tương quan tuyến tính trong một phạm vi rộng với sự phong phú của protein, giúp cho phép đo của chúng khả thi đối với các nghiên cứu định lượng5.
Định lượng dựa không có nhãn so với có nhãn
Đối với phân tích không nhãn thông qua cường độ ion peptide, nên sử dụng máy quang phổ khối có độ phân giải cao vì khối lượng của các ion tiền chất cần được xác định rất chính xác. Phân tích không nhãn bằng cách đếm phổ cũng có thể được thực hiện trên máy quang phổ khối có độ phân giải thấp và cho thấy kết quả định lượng chính xác hơn so với phương pháp dựa trên cường độ ion6. Phương pháp không nhãn có phạm vi hoạt động lớn nhất và độ phủ proteome cao nhất để nhận dạng. Tuy nhiên, độ chính xác định lượng và khả năng tái tạo kém hơn so với các chiến lược dựa trên nhãn đã nghiên cứu7. Phương pháp không nhãn cũng được phát hiện là mang lại độ phủ proteome cao hơn. Trong chính phương pháp không nhãn, những khác biệt đáng kể cũng được theo dõi tùy thuộc vào phần mềm được sử dụng để phân tích dữ liệu.
Một lợi thế rõ ràng của các chiến lược dựa trên nhãn so với các phương pháp không nhãn là khả năng phân tích ghép kênh, cho phép đo đồng thời các mẫu được gắn nhãn khác biệt trong một thí nghiệm duy nhất. Đặc biệt, khả năng ghép kênh của 2-plex, 4-plex và 8-plex có thể đạt được bằng các thuốc thử iTRAQ và TMT có bán trên thị trường. Nhưng các chiến lược gắn nhãn chuyển hóa không áp dụng được cho phân tích proteome của các mẫu lâm sàng và do đó bị hạn chế đối với loại mẫu. Mặt khác, các chiến lược gắn nhãn hóa học như iTRAQ hoặc TMT có các yêu cầu đặc biệt liên quan đến máy quang phổ khối bẫy ion. Ngược lại với các phương pháp không có nhãn, iTRAQ hoặc TMT yêu cầu các phương pháp phân mảnh thay thế như HCD (phân ly va chạm năng lượng cao hơn) hoặc ETD (phân ly chuyển điện tử) nên được sử dụng thay cho CID (phân ly va chạm gây ra), không tương thích với các ion báo cáo phạm vi khối lượng thấp1.
Bảng 1 – Định lượng dựa trên nhãn so với định lượng không nhãn
| Định lượng dựa trên nhãn (iTRAQ/TMT) | Định lượng không cần nhãn | |
| Thông lượng (số lượng nhận dạng protein) | Cao | Thấp |
| Số lượng mẫu được phát hiện | Số lượng thẻ có hạn | Ít hạn chế hơn, phù hợp với kích thước mẫu lớn |
| Số lượng cần thiết cho mẫu ban đầu | Cao | Thấp |
| Phát hiện sự có mặt hoặc vắng mặt của protein | — | Có thể phát hiện |
| Yêu cầu song song mẫu | Tính song song tốt, khả năng tái tạo thí nghiệm cao | Yêu cầu cao về tính song song và khả năng tái tạo các thí nghiệm thấp |
| Yêu cầu mẫu | Nền mẫu tương tự (cùng loài và cùng loại mẫu) | Các mẫu từ các loài, nguồn hoặc địa điểm khác nhau yêu cầu phân tích protein đồng thời |
| Lỗi thử nghiệm | Việc dán nhãn và nhóm pha lỏng có thể gây ra lỗi thực nghiệm | Không có lỗi nào được giới thiệu |
| Omics đã sửa đổi | Chỉ phù hợp cho các dự án sửa đổi phosphoryl hóa | Có thể sửa đổi tổng quát omics |
Các khía cạnh thực nghiệm
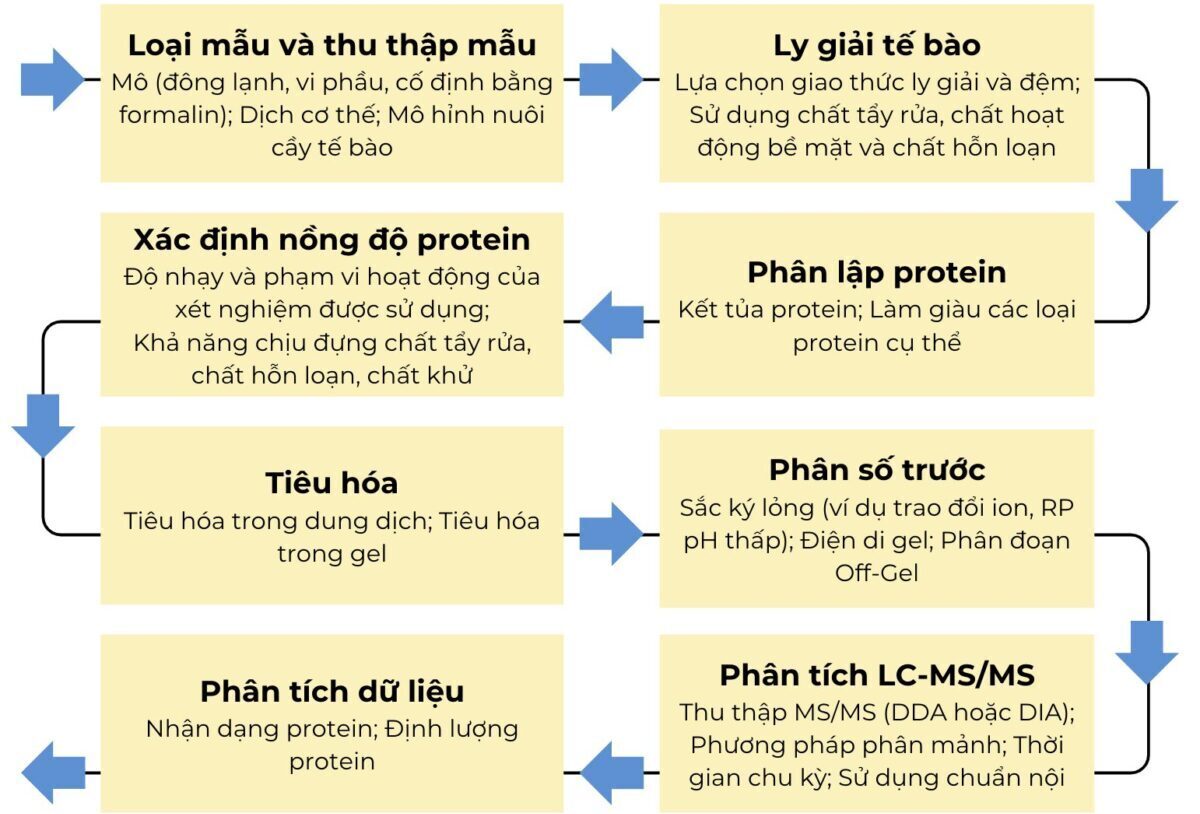
Hình 1 – Quy trình làm việc tổng quát của phân tích proteome định lượng không nhãn. Quy trình làm việc bao gồm các bước phân tích dữ liệu và thử nghiệm có thể ảnh hưởng đến kết quả của nghiên cứu proteome định lượng không nhãn và có khả năng gây ra một số sai lệch.
a. Chuẩn bị mẫu, tinh chế và tách
Tính mạnh mẽ và khả năng tái tạo của việc chuẩn bị mẫu là hai khía cạnh quan trọng nhất đối với một phân tích proteome định lượng thành công, đặc biệt là đối với định lượng không có nhãn. Do đó, mọi bước trong quy trình thực nghiệm đều có khả năng gây ra lỗi và có thể đưa ra một số sai lệch có thể tạo ra kết quả gây hiểu lầm. Bước đầu tiên của phân tích proteome định lượng đối với các mẫu lâm sàng quyết định quy trình làm việc đang diễn ra là lựa chọn loại mẫu và cách thu thập mẫu8. Các quy trình làm việc được tối ưu hóa để phân tích DIGE 2D định lượng của các tế bào được vi phẫu có nguồn gốc từ nhiều mẫu mô khác nhau đã được phát triển và áp dụng thành công trong một số nghiên cứu phát hiện ra dấu ấn sinh học9, 10. Các bước tiếp theo, cụ thể là ly giải tế bào, phân lập protein và tiêu hóa, cũng có thể ảnh hưởng đáng kể đến kết quả của một nghiên cứu proteomics. Cần lựa chọn các đệm ly giải tế bào và điều kiện khác nhau tùy thuộc vào phần proteome đang được nghiên cứu (ví dụ: protein màng, protein bào quan, protein tế bào chất) hoặc chính loại mẫu. Ngoài các giao thức ly giải, các giao thức tiêu hóa cũng phải được lựa chọn cẩn thận. Ngay cả khi tiêu hóa bằng phương pháp phân giải protein với trypsin hoặc các protease khác là phương pháp được sử dụng rộng rãi nhất trong nghiên cứu về protein, các phương pháp tiêu hóa dựa trên phương pháp hóa học hoặc vật lý có thể hữu ích và cần thiết trong những trường hợp đặc biệt. Nếu sử dụng chất tẩy rửa có thể ảnh hưởng đến hiệu suất của sắc ký lỏng trong quá trình ly giải tế bào, thì nên sử dụng chất tẩy rửa trước vì nó như một bước tinh chế nội tại của protein và peptide trong gel. Tuy nhiên, phương pháp này cũng mất mẫu đáng kể do không chiết xuất hoàn toàn các peptide từ một mảnh gel. Ngoài ra, các cột spin ngày càng được sử dụng để tinh chế mẫu11 và các giao thức tiêu hóa trên bộ lọc tương ứng cũng khả dụng12.
Ngoài các bước đã đề cập ở trên, việc xác định nồng độ protein trong một mẫu nhất định là một khía cạnh thực nghiệm rất quan trọng, đặc biệt là trong trường hợp định lượng không có nhãn. Cần xác định trực tiếp sau khi chiết xuất protein từ mẫu để xác nhận việc phân lập thành công protein và điều chỉnh lượng protease được sử dụng trong bước tiêu hóa sau. Tuy nhiên, nếu tiêu hóa bằng gel, việc xác định thêm nồng độ peptide sau khi chiết xuất từ gel sẽ có lợi, vì hiệu suất chiết xuất thay đổi có thể dẫn đến các quy định peptide nhân tạo trong phân tích không nhãn tiếp theo. Một số khía cạnh cần được xem xét để lựa chọn xét nghiệm phù hợp là: phạm vi làm việc và giới hạn phát hiện của xét nghiệm, khả năng chịu chất tẩy rửa, chất hỗn loạn hoặc chất khử và câu hỏi liệu có nên nghiên cứu peptide hay protein hay không. Đối với phân tích định lượng không nhãn theo cường độ peptide ion, khả năng tái tạo của bước này cực kỳ quan trọng vì khối lượng tiền chất peptide được đo được khớp với thời gian lưu tương ứng của chúng. Để đảm bảo khả năng tái tạo sắc ký trong một nghiên cứu không nhãn, việc sử dụng một chuẩn nội bao gồm các peptide được mã hóa bằng đồng vị ổn định là có lợi. Nếu muốn hoặc cần thiết, mẫu có thể được phân đoạn trước (ví dụ thông qua 1D-PAGE, tập trung đẳng điện, sắc ký trao đổi ion, sắc ký lỏng pha đảo pH cao) trước RP-LC và sau đó mỗi phân đoạn có thể được phân tích trong một thí nghiệm LC–MS/MS duy nhất. Một lợi thế rõ ràng của thiết lập hai chiều như vậy là sự phân tách phức tạp của mẫu dẫn đến độ phủ proteome cao hơn cũng như khả năng chọn các phân đoạn cụ thể quan tâm cho các phân tích tiếp theo13, 14. Tuy nhiên, để có được kết quả định lượng đáng tin cậy và tránh bất kỳ sai lệch nào, mỗi bước của quá trình phân đoạn trước phải được thực hiện theo cách có khả năng tái tạo cao. Hơn nữa, cần lưu ý rằng số lượng thí nghiệm LC–MS/MS tăng đáng kể theo số lượng phân đoạn đã tách trước đó.
b. Phân tích phổ khối
Tùy thuộc vào máy quang phổ khối được sử dụng để phân tích không nhãn, có thể sử dụng các chế độ thu thập khác nhau. Nhìn chung, các phân tích khối phổ để định lượng không nhãn có thể được thực hiện theo hai cách khác nhau:
- Thu thập phụ thuộc dữ liệu (DDA):
- Bao gồm việc thu thập quét khảo sát và phân mảnh tiếp theo các ion peptide tiền chất đã chọn. Ở đây, người ta nên nhớ rằng cần phải tìm ra sự cân bằng phù hợp giữa phổ khảo sát thu được và phổ ion mảnh. Ví dụ, số lượng phổ phân mảnh thu được tăng lên dẫn đến độ bao phủ proteome cao hơn, nhưng đồng thời làm tăng thời gian chu kỳ của toàn bộ quá trình thu thập MS/MS. Điều này lần lượt dẫn đến ít phổ khảo sát MS thu được hơn, cần thiết để mô tả đỉnh ion peptide sắc ký để định lượng. Do đó, các thông số phổ khối như số phổ ion mảnh thu được, thời gian thu MS, phương pháp phân mảnh và nhiều thông số khác phải được điều chỉnh và tối ưu hóa cẩn thận đối với hành vi sắc ký của mẫu để tạo ra kết quả định lượng đáng tin cậy và có thể tái tạo. Loại thu thập này cho phép định lượng thông qua đếm phổ cũng như đo cường độ ion peptide1.
- Định lượng proteomics MS1 không có nhãn dựa trên DDA:
- Mỗi mẫu đầu tiên được cắt bằng enzym thành một peptide. Sau đó, các mẫu được tiến hành phân tích LC-MS/MS bằng phương pháp thu thập dữ liệu phụ thuộc (DDA). Quá trình quét DDA bao gồm quét toàn bộ MS1 có độ phân giải cao, tiếp theo là bẫy ion/thanh bốn lần để phân lập các ion mẹ cho các điều kiện cụ thể, phân mảnh tế bào va chạm và thu thập phổ ion phân mảnh MS2. Sắc ký dòng ion của ion mẹ có độ phân giải cao đã chiết xuất được sử dụng làm đặc điểm định lượng và MS2 được sử dụng để nhận dạng trình tự peptide.
- Phương pháp này dựa trên cường độ ion mẹ phổ chính, chẳng hạn như chiều cao đỉnh phổ, diện tích đỉnh phổ và thông tin về thể tích đỉnh phổ để định lượng protein. Trong phổ khối chính, mỗi ion mẹ là một peptide ion hóa, bao gồm thông tin ba chiều như thời gian lưu sắc ký lỏng, tỷ lệ khối lượng trên điện tích và cường độ ion. Cường độ tín hiệu ion mẹ có tương quan với nồng độ ion. Do đó, cường độ đỉnh ion (chiều cao đỉnh, diện tích đỉnh và thể tích đỉnh, v.v.) tương ứng với peptide đã xác định được trích xuất từ phổ chính để phản ánh sự phong phú của peptide.
- Dựa trên chiến lược này, định lượng MS1 và nhận dạng MS2 độc lập với nhau, cho phép thông tin nhận dạng peptide được truyền qua toàn bộ tập dữ liệu mẫu và tạo điều kiện định lượng peptide có hàm lượng thấp. Chiến lược định lượng MS1 thường được sử dụng cho các nghiên cứu proteomics định lượng quy mô lớn.
- Thu thập không phụ thuộc dữ liệu (DIA):
- Máy quang phổ khối liên tục hoạt động ở chế độ MS/MS. Ngược lại với DDA, không có lựa chọn ion tiền chất nào được thực hiện trong quá trình quét khảo sát MS. Thay vào đó, dữ liệu khối được thu thập bằng cách xen kẽ năng lượng va chạm giữa trạng thái năng lượng thấp và cao. Phương pháp này được gọi là LC–MSE (E là cao) và cho thấy tỷ lệ nhiễu tín hiệu tăng lên và có thể dẫn đến tỷ lệ nhận dạng tốt hơn các peptide có hàm lượng thấp có thể bị bỏ sót trong quá trình phân tích phụ thuộc vào dữ liệu1.
- Phân tích định lượng không nhãn dựa trên DIA:
- Thu thập dữ liệu độc lập (DIA) được thực hiện bằng cách phân mảnh MS/MS tất cả các ion cha mẹ peptide trong phạm vi tỷ lệ khối lượng trên điện tích (m/z) cụ thể một cách ngẫu nhiên sau khi quét toàn bộ khối phổ chính có độ phân giải cao. Trong DIA, phổ MS2 có độ phân giải cao được sử dụng để nhận dạng peptide và cả MS1 và MS2 có độ phân giải cao đều có thể được sử dụng để định lượng peptide/protein.
- So với phương pháp định lượng dựa trên MS1, ưu điểm của phương pháp DIA là (1) tiếp cận không phân biệt đối xử với tất cả các peptide mà không làm mất thông tin của các protein có hàm lượng thấp, thể hiện qua ít giá trị bị thiếu hơn; (2) thời gian chu kỳ cố định, số điểm quét đồng đều và độ chính xác định lượng cao, thể hiện qua hệ số biến thiên (CV) thấp hơn trong cường độ của các protein được định lượng; (3) hệ số CV thấp hơn); (3) không có tính ngẫu nhiên trong việc lựa chọn peptide và dữ liệu có thể được truy xuất lại; (4) khả năng tái tạo tốt hơn đối với các mẫu protein phức tạp, đặc biệt là các protein có hàm lượng thấp.
c. Các nghiên cứu về proteomics lâm sàng không nhãn
Từ khi proteomics không nhãn được giới thiệu, nhiều nghiên cứu đa dạng nhằm mục đích khám phá các dấu ấn sinh học hoặc mục tiêu thuốc đã được thực hiện bằng các phương pháp tiếp cận không nhãn.
Yangetal đã thực hiện một phân tích so sánh không nhãn của bệnh nhân ung thư đại tràng tập trung vào các protein tế bào xương và các đối tác tương tác của chúng cũng như các protein ma trận ngoại bào. Họ đã tìm thấy 56 protein được biểu hiện khác biệt giữa các mẫu mô từ những người khỏe mạnh và các mẫu khối u15. Gần đây, nhóm của chúng tôi đã mô tả một nghiên cứu không nhãn kết hợp với phương pháp tiếp cận 2D-DIGE, nghiên cứu ung thư biểu mô tế bào gan (HCC). Sáu protein ứng viên đã được chọn và xác minh bằng các phương pháp miễn dịch học sử dụng cùng các mẫu được sử dụng để phát hiện. Hai protein được phát hiện có sự biểu hiện khác biệt đáng kể và do đó đại diện cho các ứng cử viên sinh học mới cho HCC16.
Việc sử dụng các mẫu cố định bằng formalin để phân tích không có nhãn cũng đã được báo cáo. Hyung và cộng sự đã báo cáo một nghiên cứu về proteomics không có nhãn khác biệt sử dụng các mẫu huyết thanh người. Họ phân đoạn các mẫu huyết thanh của bệnh nhân ung thư vú bằng cách phân lập các protein N-glycosylated bằng hóa học hydrazide do đó tránh làm cạn kiệt các protein có độ phong phú cao trước khi phân tích17. Mặc dù làm suy giảm miễn dịch được sử dụng rộng rãi, nhưng người ta nên lưu ý đến những bất lợi của quy trình này khi làm việc với các mẫu huyết thanh hoặc huyết tương. Làm suy giảm miễn dịch khó có thể tái tạo và rất có thể gây ra sai lệch trong quá trình chuẩn bị mẫu. Điều tương tự cũng đúng đối với quá trình đông máu, nhưng hơn nữa, một số loại protease cũng tham gia vào quá trình đông máu. Vì lý do này, nên tránh làm suy giảm miễn dịch nếu có thể và huyết tương là mẫu được lựa chọn khi các mẫu máu được nghiên cứu bằng các nghiên cứu về protein định lượng.
Các mô hình nuôi cấy tế bào đưa ra một cách lý tưởng để nghiên cứu các câu hỏi sinh học trong một hệ thống được xác định rõ ràng với sự biến đổi sinh học ít hơn. Mặt khác, điều này cũng có nghĩa là sự biểu diễn thiếu hụt của sự đa dạng sinh học phức tạp mà người ta phải xem xét khi giải quyết các câu hỏi có liên quan đến lâm sàng. Tuy nhiên, một số nghiên cứu không nhãn sử dụng các mô hình nuôi cấy tế bào để giải quyết các câu hỏi như vậy đã được báo cáo.
Tài liệu tham khảo:
1. D. A. Megger, T. Bracht, H. E. Meyer, and B. Sitek. Label-free quantification in clinical proteomics, Biochim Biophys Acta 2013, 1834(8), 1581-90.
2. H. Liu, R. G. Sadygov, and J. R. Yates, 3rd. A model for random sampling and estimation of relative protein abundance in shotgun proteomics, Anal Chem 2004, 76(14), 4193-201.
3. P. Lu, C. Vogel, R. Wang, X. Yao, and E. M. Marcotte. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation, Nature Biotechnology 2007, 25(1), 117-124.
4. L. Florens, M. J. Carozza, S. K. Swanson, M. Fournier, M. K. Coleman, J. L. Workman, and M. P. Washburn. Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors, Methods 2006, 40(4), 303-11.
5. P. V. Bondarenko, D. Chelius, and T. A. Shaler. Identification and relative quantitation of protein mixtures by enzymatic digestion followed by capillary reversed-phase liquid chromatography-tandem mass spectrometry, Anal Chem 2002, 74(18), 4741-9.
6. N. M. Griffin, J. Yu, F. Long, P. Oh, S. Shore, Y. Li, J. A. Koziol, and J. E. Schnitzer. Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis, Nat Biotechnol 2010, 28(1), 83-9.
7. Z. Li, R. M. Adams, K. Chourey, G. B. Hurst, R. L. Hettich, and C. Pan. Systematic comparison of label-free, metabolic labeling, and isobaric chemical labeling for quantitative proteomics on LTQ Orbitrap Velos, J Proteome Res 2012, 11(3), 1582-90.
8. R. E. Banks, M. J. Dunn, M. A. Forbes, A. Stanley, D. Pappin, T. Naven, M. Gough, P. Harnden, and P. J. Selby. The potential use of laser capture microdissection to selectively obtain distinct populations of cells for proteomic analysis–preliminary findings, Electrophoresis 1999, 20(4-5), 689-700.
9. C. Molleken, B. Sitek, C. Henkel, G. Poschmann, B. Sipos, S. Wiese, B. Warscheid, C. Broelsch, M. Reiser, S. L. Friedman, I. Tornoe, A. Schlosser, G. Kloppel, W. Schmiegel, H. E. Meyer, U. Holmskov, and K. Stuhler. Detection of novel biomarkers of liver cirrhosis by proteomic analysis, Hepatology 2009, 49(4), 1257-66.
10. G. Poschmann, B. Sitek, B. Sipos, and K. Stuhler. Application of saturation labeling in lung cancer proteomics, Methods Mol Biol 2012, 854, 253-67.
11. B. S. Antharavally, K. A. Mallia, M. M. Rosenblatt, A. M. Salunkhe, J. C. Rogers, P. Haney, and N. Haghdoost. Efficient removal of detergents from proteins and peptides in a spin column format, Anal Biochem 2011, 416(1), 39-44.
12. J. R. Wisniewski, A. Zougman, N. Nagaraj, and M. Mann. Universal sample preparation method for proteome analysis, Nat Methods 2009, 6(5), 359-62.
13. L. M. Blumberg, F. David, M. S. Klee, and P. Sandra. Comparison of one-dimensional and comprehensive two-dimensional separations by gas chromatography, J Chromatogr A 2008, 1188(1), 2-16.
14. V. Gautier, E. Mouton-Barbosa, D. Bouyssie, N. Delcourt, M. Beau, J. P. Girard, C. Cayrol, O. Burlet-Schiltz, B. Monsarrat, and A. Gonzalez de Peredo. Label-free quantification and shotgun analysis of complex proteomes by one-dimensional SDS-PAGE/NanoLC-MS: evaluation for the large scale analysis of inflammatory human endothelial cells, Mol Cell Proteomics 2012, 11(8), 527-39.
15. H. Y. Yang, J. Kwon, H. R. Park, S. O. Kwon, Y. K. Park, H. S. Kim, Y. J. Chung, Y. J. Chang, H. I. Choi, K. J. Chung, D. S. Lee, B. J. Park, S. H. Jeong, and T. H. Lee. Comparative proteomic analysis for the insoluble fractions of colorectal cancer patients, J Proteomics 2012, 75(12), 3639-53.
16. D. A. Megger, T. Bracht, M. Kohl, M. Ahrens, W. Naboulsi, F. Weber, A. C. Hoffmann, C. Stephan, K. Kuhlmann, M. Eisenacher, J. F. Schlaak, H. A. Baba, H. E. Meyer, and B. Sitek. Proteomic differences between hepatocellular carcinoma and nontumorous liver tissue investigated by a combined gel-based and label-free quantitative proteomics study, Mol Cell Proteomics 2013, 12(7), 2006-20.
17. S. W. Hyung, M. Y. Lee, J. H. Yu, B. Shin, H. J. Jung, J. M. Park, W. Han, K. M. Lee, H. G. Moon, H. Zhang, R. Aebersold, D. Hwang, S. W. Lee, M. H. Yu, and D. Y. Noh. A serum protein profile predictive of the resistance to neoadjuvant chemotherapy in advanced breast cancers, Mol Cell Proteomics 2011, 10(10), M111 011023.
Uyen Nguyen (Research Officer, Hoan Vu Biomolecules., JSC.)
BÀI VIẾT MỚI >>
- Các phương pháp phân tích định lượng, định tính
 Các phương pháp phân tích định lượng, định tính Giá trên đã bao gồm thuế phí.
Các phương pháp phân tích định lượng, định tính Giá trên đã bao gồm thuế phí. - Các phương pháp phân tích Sinh Hóa Lý
- Tổng quan về ELISA
 ELISA (Enzyme-linked immunosorbent assay) là kỹ thuật xét nghiệm miễn dịch sử dụng enzyme để phát hiện và định lượng kháng nguyên hoặc kháng thể. Có […]
ELISA (Enzyme-linked immunosorbent assay) là kỹ thuật xét nghiệm miễn dịch sử dụng enzyme để phát hiện và định lượng kháng nguyên hoặc kháng thể. Có […] - Các phương pháp chiết tách protein trong nghiên cứu proteomics và ứng dụng
 Các tiến bộ trong công nghệ proteomics không thể khắc phục được các vấn đề trong chuẩn bị mẫu. Các bước như đồng nhất hóa mô, […]
Các tiến bộ trong công nghệ proteomics không thể khắc phục được các vấn đề trong chuẩn bị mẫu. Các bước như đồng nhất hóa mô, […] - Sự hình thành cầu disulfide trong protein
 Tầm quan trọng của cầu nối Disulfide Ổn định cấu trúc Protein: Cầu nối disulfide giúp ổn định cấu trúc bậc ba và bậc bốn của […]
Tầm quan trọng của cầu nối Disulfide Ổn định cấu trúc Protein: Cầu nối disulfide giúp ổn định cấu trúc bậc ba và bậc bốn của […]


